| In
Vitro Anti-Hepatitis B Virus Activities of 5’-O-Myristoyl Analogue Derivatives
of 3’-Fluoro-2’,3’-dideoxythymidine (FLT) and
3’-Azido-2’,3’-dideoxythymidine (AZT) Manuscript received September 30th, 1998; Reviewed October 28th, 1998; Accepted November 26th, 1998.Keykavous Parang, Leonard I. Wiebe, Edward E. Knaus1
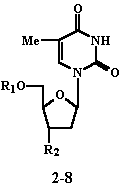
Jyy-Shiang Huang, David L. Tyrrell Abstract Purpose: The objective of this study was to evaluate a dual action prodrug concept wherein an unnatural myristic acid analogue is coupled via an ester moiety to the 5’-position of FLT or AZT. Subsequent intracellular cleavage of the prodrug ester would simultaneously release FLT or AZT that could inhibit reverse transcriptase (RT), and the myristic acid analogue that could inhibit myristoyl-CoA:protein N-myristoyltransferase (NMT). Methods: Cytotoxicity (2.2.15 cell culture), and anti-hepatitis B activity of 5’-O-myristoyl analogue prodrug derivatives of FLT and AZT (2-8) were evaluated in vitro using human liver hepatitis B virus (HBV) producing 2.2.15 cell lines. Results: The 5’-O-(12-methoxydodecanoyl) ester derivatives of AZT (2, EC50 = 2.7 ± 0.3 mM; CC50 = 727 ± 19 mM) and FLT (4, EC50 = 2.8 ± 0.3 mM; CC50 = 186 ± 20 mM) were the most effective anti-hepatitis B virus (anti-HBV) compounds of this series in a replication assay. In the series of 5’-O-myristic acid analogue ester prodrug derivatives of FLT, the relative anti-HBV potency order was MeO(CH2)11CO2- > N3(CH2)11CO2- and Br(CH2)11CO2- > EtS(CH2)nCO2- (n = 10 or 11) > Me(CH2)12CO2- (myristoyl). Conclusions: The in vitro data suggest that the 5’-O-myristoyl analogue prodrug concept offers a potential drug design approach to design dual acting anti viral agents, with superior pharmacokinetic, biodistribution, reduced cytotoxicity and/or increased efficacy. In this regard, the 5’-O-(12-methoxydodecanoyl) prodrug ester of 3’-thia-3’-deoxythymidine (3TC) may offer the greatest potential for the treatment of HBV infection. Introduction Five percent of the world population, or over 300 million people, are chronically infected with hepatitis B virus (HBV) (1). Troisi et al. (2) reported that 75% of human immunodeficiency virus (HIV) seropositive men also showed evidence of HBV infection. One notable similarity between these two human pathogenic viruses is that their replicative cycles involve an obligate RNA intermediate and a reverse transcription step in the cytoplasm. HBV contains a specific DNA polymerase, that is vital for replication, which acts as a reverse transcriptase during the replication of HBV DNA via an RNA intermediate (3). Although vaccination has been used for the prevention of HBV infection, there are no clinically effective drugs available for the treatment of HBV infection. Recently, (-)-b -2’,3’-dideoxy-3’-thiacytidine (3TC), which has been the subject of clinical trials in HBV-infected patients (4), and 2’,3’-dideoxycytidine (ddC) were shown to be potent inhibitors of hepatitis B virus replication in vitro (5). However, long-term ddC administration causes delayed toxicity such as peripheral neuropathy in patients, which was attributed to depletion of mitochondrial DNA in host cells (6). Since HBV causes severe and often fatal side effects, there is an urgent need to design novel and effective drugs with low host toxicity. The in vitro anti-HBV activities for 5’-O-myristoyl analogue derivatives of AZT and FLT (2-8) were investigated in this study (Tables 1 and 2).
These ester prodrugs were designed with the expectation that two anti-HBV agents (nucleoside and myristic acid analogue) would be released upon ester cleavage of the prodrug. The parent nucleoside (FLT or AZT) and the myristic acid analogue have different biochemical targets thereby increasing the probability of enhanced anti-HBV efficacy. This approach includes inhibition of reverse transcriptase by FLT and AZT, and inhibition of myristoyl-CoA:protein N-myristoyltransferase (NMT) that catalyzes the myristoylation of viral proteins by the myristic acid analogue (7). Myristoylation by unnatural myristic acid analogues may block the synthesis of the PreS1 protein in the membrane envelope of HBV (8). 12-Methoxydodecanoic acid (1), a myristic acid analogue, was previously reported to be a moderately active inhibitor of HBV DNA replication in 2.2.15 cell culture [HBV DNA replication intermediates (HBV RI intracellular DNA), HBV virion extracellular DNA] compared to the reference compound ddC and it was less toxic (9). Although myristoylation-null HBV mutants form virion particles, these particles are non-infectious (8).
Although AZT and FLT exhibit in vitro anti-HIV activity (10), data from clinical trials in HIV-seropositive patients suggested that FLT induced severe toxic effects such as thrombocytopenia and anemia, providing a narrow therapeutic window (11) that resulted in the termination of clinical trials. 5’-O-Myristoyl analogue prodrugs of FLT may have a number of potential advantages relative to FLT, including (i) a reduction in the toxicity of FLT, (ii) two different mechanisms of action for greater therapeutic efficacy, and (iii) the myristic acid analogue moiety does not induce bone marrow toxicity. Further, to treat hepatitis B more effectively, it would be desirable to deliver a larger percentage of the antiviral nucleoside dose to the liver. It was anticipated that the lipophilic 5’-O-myristic acid analogue prodrugs (2-8) would undergo extensive hepatic uptake since the non-polar myristoyl analogue ester moiety increases lipophilicity. The parent nucleosides, AZT and FLT, also exhibit in vitro anti-HIV activity (10). Since multiple concomitant viral infections frequently occur, there would be a potential clinical benefit to treat both HBV and HIV infections using a single drug. FLT is one of the most effective antiviral agents in vitro, producing a greater than 90% reduction (0.03 mM) in HBV DNA synthesis. FLT 5’-triphosphate is a potent inhibitor of endogenous HBV DNA polymerase, preventing the formation of HBV in HepG2 cells transfected with HBV DNA in vitro (12). In contrast, AZT failed to suppress the replication of HBV in patients with HIV and HBV infection (13-15) and was relatively ineffective against HBV replication in 2.2.15 cells even at the highest possible concentration (20 mM) that did not affect cellular growth (5, 16). AZT had no appreciable effects on HBV DNA replication in HIV patients with chronic hepatitis B infection, during long term therapy (13) and it did not inhibit HBV DNA polymerase as determined from in vitro assays. Since AZT is metabolized in the liver (17), effective levels of the active triphosphate may not be present in hepatocytes even though AZT-triphosphate (AZT-TP) inhibits HBV DNA polymerase in vitro (EC50 = 0.15 mM) (18). The action of AZT against HBV appears to be related to inhibition of HBV DNA polymerase (18). Materials and Methods Chemistry 12-Methoxydodecanoic acid (1) (7), 3’-azido-2’,3’-dideoxythymidine (AZT) (19) and 3’-fluoro-2’,3’-dideoxythymidine (FLT) (10) were prepared according to literature methods. The 5’-OH of AZT (1.3 mmol) was esterified using 12-methoxydodecanoic acid (1) (1.3 mmol) in anhydrous benzene (18 mL) in the presence of oxalyl chloride (1.9 mmol) and 4-(dimethylamino)pyridine (DMAP) (1.9 mmol) to yield 3’-azido-2’,3’-dideoxy-5’-O-(12-methoxydodecanoyl)thymidine (2) in 56% yield (20). A similar procedure was used for the preparation of the 5’-O-myristoyl analogue derivatives of FLT (3-7) (21). Further reaction of 3’-fluoro-2’,3’-dideoxy-5’-O-(12-bromododecanoyl)thymidine (3) with sodium azide at room temperature in dimethyl formamide produced the corresponding 12-azido analog (8) (21). In Vitro Anti-HBV Assays Two assay methods were used to determine anti-HBV activity. These two assays differ in the duration of exposure, harvesting time, and the method used to detect HBV DNA (direct beta scanner in NIH method versus dot blot hybridization in our laboratory). The first assay (results shown in Tables 1 and 2) used to measure anti-HBV activity in cultures of 2.2.15 cells was performed by the screening service provided by the United States National Institutes of Health (NIH) (5). Briefly, chronically HBV-producing human liver cells were seeded into 24-well tissue culture plates and grown to confluence. Test compounds were then added daily for a continuous 9 day period. Culture medium (changed daily during the treatment period) was collected and stored for analysis of extracellular (virion) HBV DNA after 0, 3, 6, and 9 days of treatment. Treated cells were lysed for 24 hours following day 9 of treatment for the analysis of intracellular HBV genomic forms. HBV DNA was then analyzed in a quantitative and qualitative manner for overall levels of HBV DNA (both extracellular and intracellular DNA) and the relative rate of HBV replication (intracellular DNA). Analysis of HBV DNA was performed using blot hybridization techniques (southern and slot blot) and [32P]-labelled HBV-specific probes. HBV DNA levels were measured by comparison to known amounts of HBV DNA standards applied to each nitrocellulose membrane (gel or slot blot). An AMBIS beta scanner, which measures the radioactive decay of the hybridized probes directly on the nitrocellulose membranes, was used for quantitative analysis. HBV DNA levels were then compared to those at Day 0 to determine the effect of drug treatment (see Table 1). The second assay used HBV-producing 2.2.15 cell cultures derived from a human hepatoblastoma cell line (Hep G2) together with the dot hybridization technique, as previously described (9). Viral replication (the values are the average of two different experiments) was estimated from the amount of HBV DNA present in the supernatant of the cell culture after incubation with the test compound (10 mg/mL, 19.8-22.0 mM range for 3-7). The data are expressed as a percentage of control values where the cell culture was not incubated with the test compound. The percentage of DNA replication in presence of the test compound was calculated using the following relationship: [Sample (treated 2.2.15 sample) - Negative Control (HepG2)] ´ 100 / [Untreated 2.2.15 Positive Control - Negative Control (HepG2)] = % replication (see Table 2). Cytotoxicity Assay Cytotoxicity induced by the test compounds in cultures of 2.2.15 cells was also determined by the United States NIH (22). Briefly, 2.2.15 cells were grown to confluence in 96 well flat-bottomed tissue culture plates and treated with test compound (in 0.2 mL culture medium/well) as described above. Four concentrations of each test compound were assayed, each in triplicate cultures, in 3 to 10-fold dilution steps. Untreated control cultures were maintained on each 96 well plate. On each 96 well plate, wells containing no cells were used to correct for light scattering. Toxicity was determined by measuring neutral red dye uptake, as determined from the absorbance at 510 nm relative to untreated cells, at 24 hours following day 9 of treatment (see Tables 1 and 2). Results and Discussion Anti-HBV activities from the first assay for selected analogues are summarized in Tables 1 (HBV virion extracellular DNA) and 2 (HBV replication intermediate intracellular DNA). The prodrug esters (2-8) were designed to act as anti-HBV agents after the release of AZT or FLT which would inhibit reverse transcriptase and, at least in part, to the anti-HBV effect of the myristic acid analogue produced after cleavage of the prodrug. 3’-Azido-2’,3’-dideoxy-5’-O-(12-methoxydodecanoyl)thymidine (2) exhibited comparable potency to 2',3'-dideoxycytidine (ddC), and the selectivity indexes (SI) were similar. Although AZT is one of the least effective nucleosides against HBV replication in 2.2.15 cells, the 5’-O-(12-methoxydodecanoyl) ester (2) showed comparable activity (EC50 = 2.7 ± 0.3 m M) to ddC (EC50 = 1.3 ± 0.1 mM) and it was less cytotoxic (CC50 = 727 ± 19 mM) than ddC (CC50 = 228 ± 2.8 mM) (Table 1). The precise basis for the increased activity of 2 relative to the inactive AZT (EC50 > 100 mM) against HBV is unknown, but it could be due to an increased intracellular concentration of 2, due to its enhanced ability to cross the cell membrane by passive difusion and/or a higher concentration of AZT produced from 2, in infected cells. Horrobin et al. (23) observed that g-linoleic acid-AZT (GLA-AZT) also inhibited herpesvirus replication, a property not found with either GLA or AZT individually. Further studies are required to determine whether the enhanced anti-HBV activity exhibited by 2 (log P = > 5) (20), relative to AZT (log P = 0.11) (21), is due to increased cellular uptake and/or facile intracellular prodrug conversion to AZT. 3’-Fluoro-2’,3’-dideoxy-5’-O-(12-methoxydodecanoyl)thymidine (4) exhibited the highest anti-HBV activity (EC50 = 5.6 mM) among the 3’-fluoro analogs tested (3-8). Although 4 was approximately equiactive to 2, it was more cytotoxic than 2 (Tables 1, 2), probably due to differences in partition coefficient, rate of intracellular ester cleavage and/or cellular uptake. It was previously reported that 12-methoxydodecanoic acid (1) exhibited weaker anti-HBV activity and was less toxic than ddC (9). This structure-activity relationship demonstrates the contribution of 12-methoxydodecanoic acid with respect to the activity exhibited by the 5’-O-12-methoxydodecanoate esters 2 and 4, which were the most potent analogues tested of the prodrugs 2-8 investigated (Tables 1, 2). Although the 5’-O-(12-methoxydodecanoyl) ester derivatives of AZT and FLT (2, 4) have similar selectivity indexes (SI = 23-43 mM) to FLT (SI = 9) in vitro, the parenteral administration of myristoyl analogue ester prodrugs of antiviral nucleosides may increase in vivo efficacy and selectivity due to improved pharmacokinetic and/or biodistribution properties that are relevant to FLT therapy for treatment of hepatitis B infection. There is credence for this assumption since it was previously reported that incubation of the myristoyl ester analogues 3-8 at 37°C with porcine liver esterase (t1/2 = 1.9-20.3 min range), rat plasma (t1/2 = 0.8-4.6 min range) and rat brain homogenate (t1/2 = 11.7-21.9 min range) possess variable enzymatic stabilities. Myristoyl ester analogues 3-8 are very lipophilic (log P = 4.3 to > 5 range) relative to AZT (log P = 0.11) and FLT (log P = -0.30) (21). The stability of 5’-O-(12-azidododecanoyl)thymidine in the cell culture medium was previously reported (24) following incubation with RPMI 1640/10% FBS at a concentration of 1 mM at 37°C. This in vitro cell culture control incubation study showed that 5’-O-(12-azidododecanoyl)thymidine was relatively stable with a half-life of eight days. Therefore, it is postulated that similar prodrugs (2-8) may exist as intact esters extracellularly in the in vitro anti-HBV assay (20, 24). The differences in anti-HBV activity exhibited by the 5’-O-myristoyl analogue derivatives of FLT (3-8) could be due to several factors such as their uptake rate into cells, their rate of intracellular conversion to FLT, and differences in the anti-HBV activity among the myristic acid analogue released after ester cleavage. Some myristoyl analog ester prodrugs (3-7) were evaluated in a second assay against HBV in 2.2.15 cell cultures derived from a human hepatoblastoma cell line (Hep G2). At a test compound concentration in the 19.8-22.0 mM range for compounds 3-7 (10 mg/mL), none of these esters significantly inhibited the replication of HBV-infected hepatocytes compared FLT or 3TC. The % of viral replication for selected compounds [compound number (% of viral replication)] was 3 (57.6%); 4 (91.2%); 5 (88.1%); 6 (87.7%), 7 (85.4%); FLT (10.8%); 3TC (0.0%) and control (100.0%). The relative anti-HBV activities of some compounds such as FLT, 3-4 and 5-6 in these two assays are different when compared to the their corresponding EC90 values in Table 1, since the harvesting time, the duration of exposure, and the time at which cells were exposed to the test compound, differ. These results indicate that evaluation in one cell culture system may not provide representative antiviral activity data. Additional studies are required to make definitive conclusions regarding the anti-HBV effects exhibited by these myristoyl prodrug compounds. The data acquired indicate that these lipophilic prodrugs of AZT and FLT may offer a method to increase in vivo efficacy and selectivity of nucleoside therapy to treat hepatitis B infection. The exact mode of action of 5’-O-myristoyl analogue derivatives of AZT and FLT will obviously remain speculative until further mechanistic investigations have been carried out. The evaluation of 5’-O-myristoyl analogue derivatives of more active nucleoside analogues such as 3TC may provide additional evidence regarding the potential value of this prodrug design concept. Acknowledgments We are grateful to the Alberta Heritage Foundation for Medical Research for a studentship award to one of us (K. Parang) and the Medical Research Council of Canada for financial support of this research. We thank the United States National Institutes of Health for providing some of the anti-HBV test results. References
Corresponding author: Edward Knaus, Faculty of Pharmacy & Pharmaceutical Sciences, University of Alberta, Canada, T6G 2N8. email:eknaus@pharmacy.ualberta.ca Key Words: Anti-HBV Activities, 5’-O-Myristoyl Analogue Prodrug Derivatives, 3’-Fluoro-2’,3’-dideoxythymidine (FLT), 3’-Azido-2’,3’-dideoxythymidine (AZT).
Published by the Canadian Society for Pharmaceutical Sciences. Copyright © 1998 by the Canadian Society for Pharmaceutical Sciences. |